
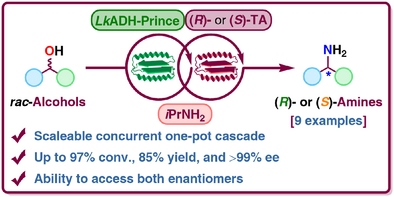
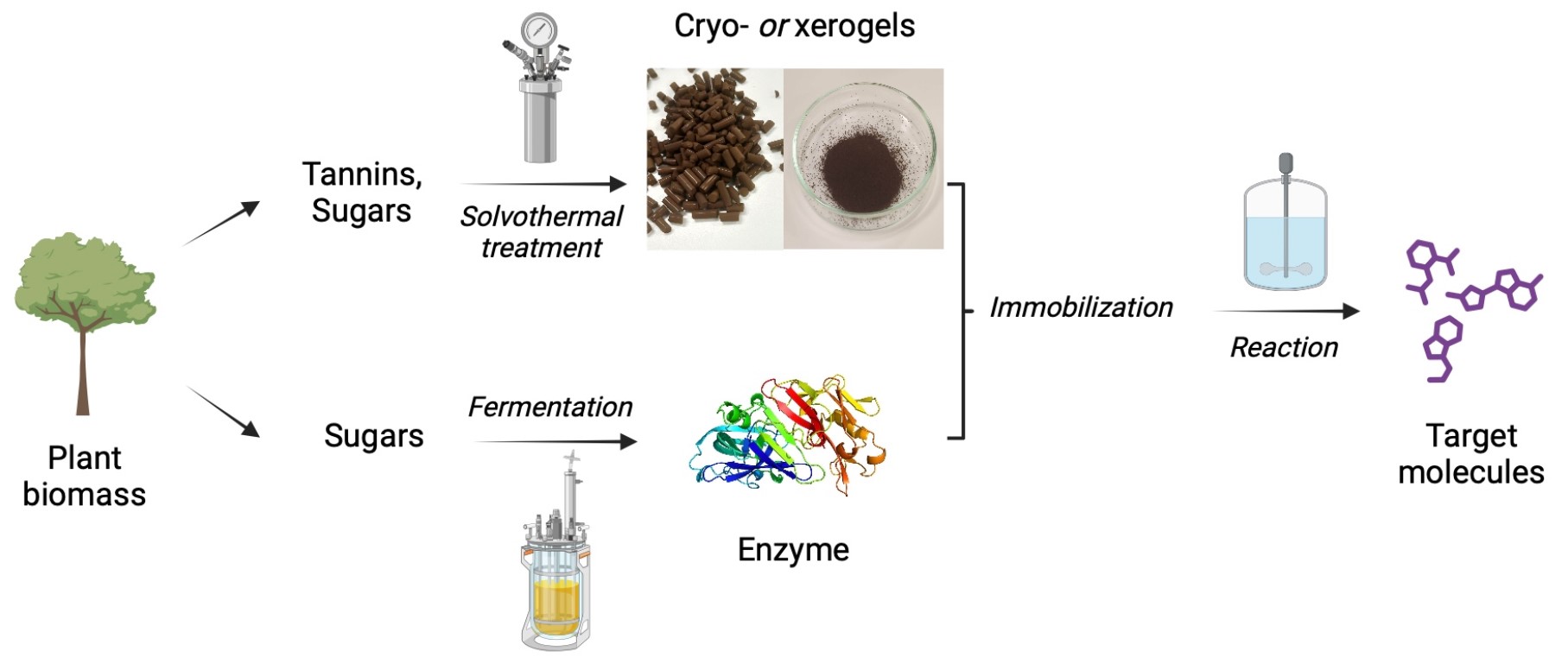
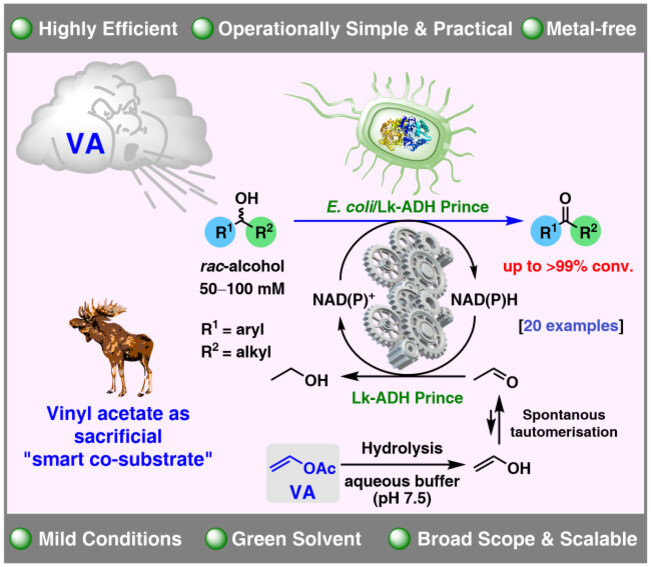
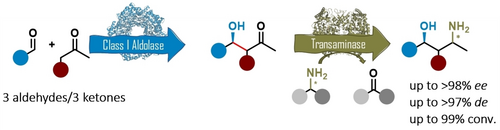
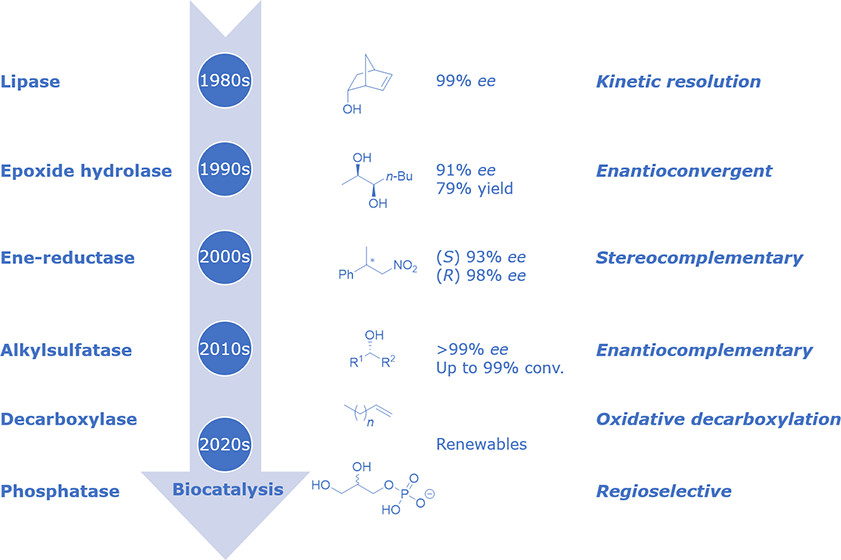
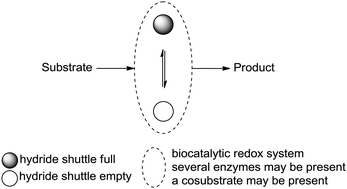
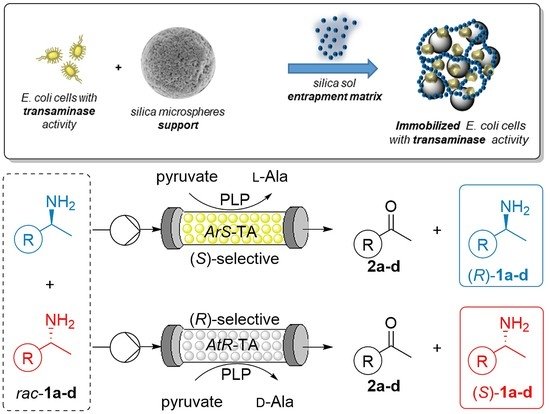
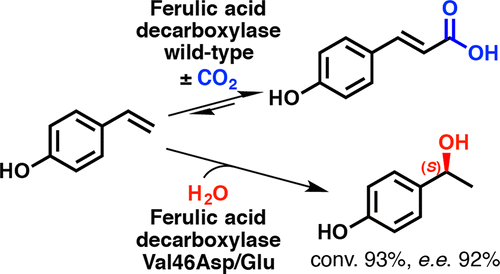
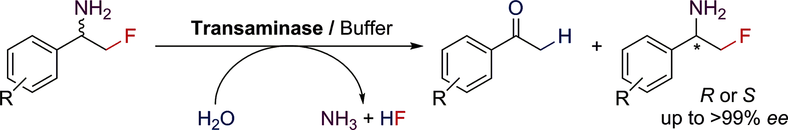
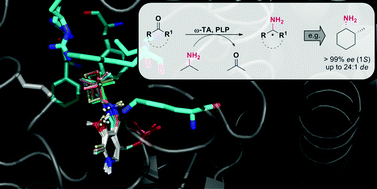
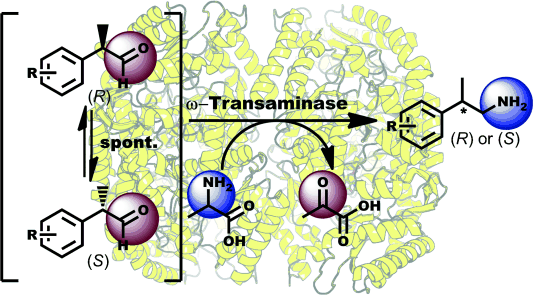
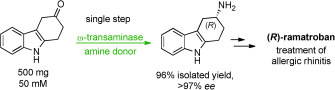
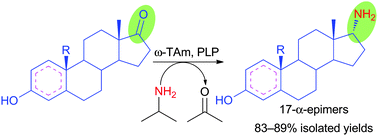
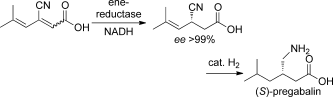
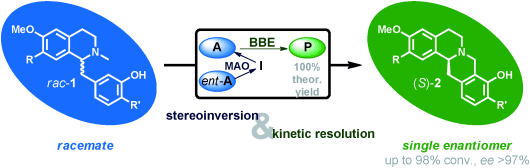
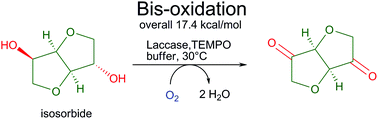
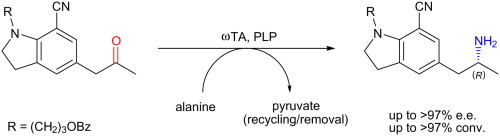
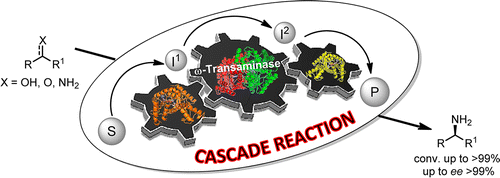
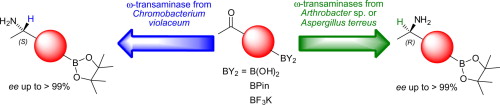
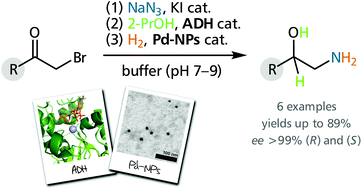
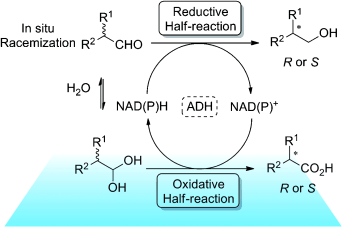
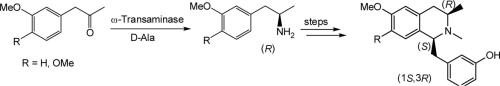
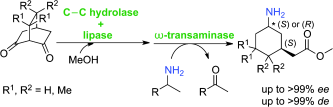
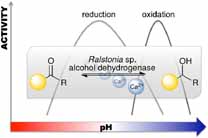
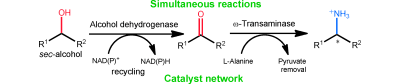
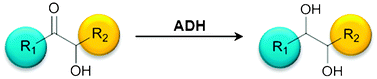
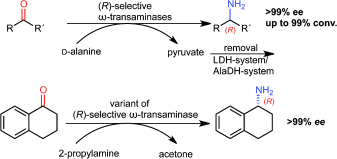
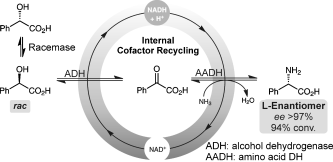
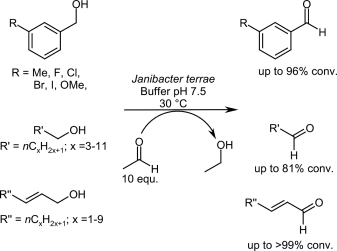
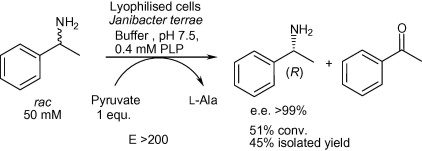
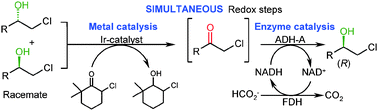
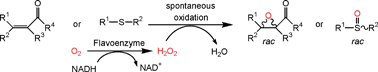
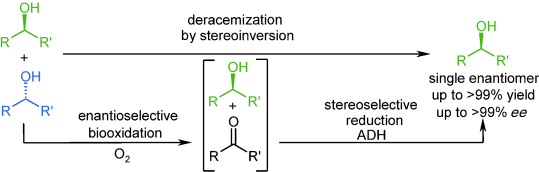
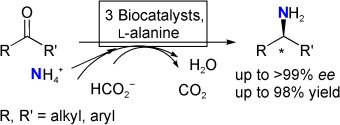
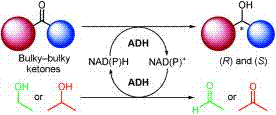
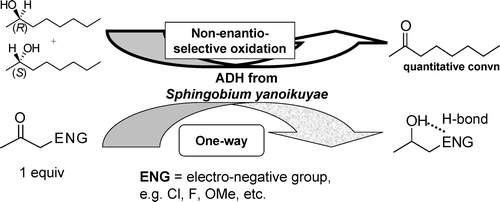
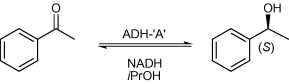
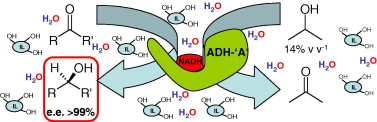
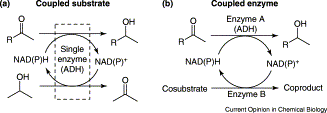
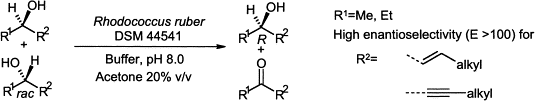
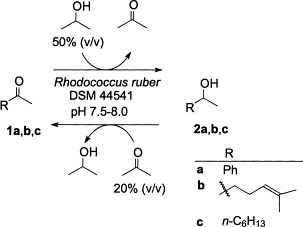
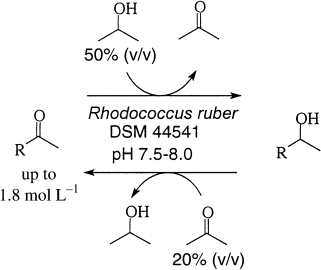
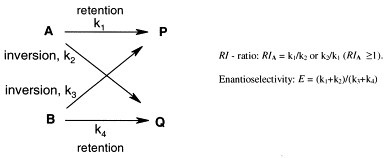
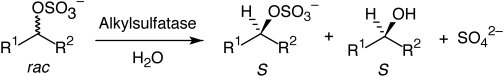
We report the development of a concurrent one-pot/two-step biocatalytic cascade for the asymmetric synthesis of enantiomerically enriched α-chiral primary amines from racemic aryl-ring-containing secondary alcohols via alcohol oxidation employing a non-enantioselective alcohol dehydrogenase, followed by asymmetric reductive amination using a stereoselective transaminase. The amine donor 2-propylamine served, on the one hand, as the nitrogen source for the amination step and, on the other hand, its deaminated/oxidized form (acetone) acted as the oxidant for the first step. The elaborated protocol employed lyophilized Escherichia coli whole cells containing a heterologously expressed ambidextrous alcohol dehydrogenase deduced from Lactobacillus kefir (E. coli/LkADH-Prince) in combination with stereocomplementary recombinant transaminases (E. coli/TAs) that simultaneously converted the in situ-generated carbonyl intermediates into the corresponding optically active amines with high-to-excellent stereocontrol. The developed cascade enables the efficient preparation of nonracemic amines with high conversions (up to 94%), very good isolated yields (up to 85%), and excellent enantiomeric excesses (up to >99%), providing access to both enantiomers under mild, nontoxic, and exclusively aqueous reaction conditions.
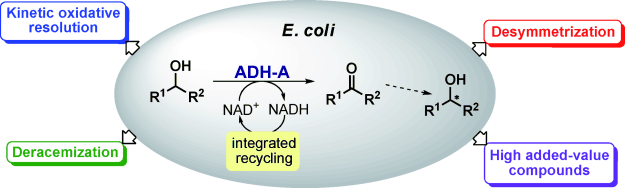
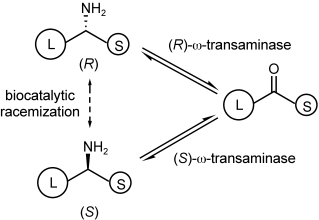
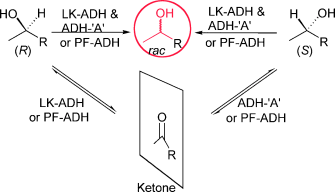
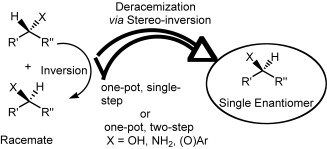
A parallel antisense one-pot/two-step concurrent cascade combining an engineered variant of an alcohol dehydrogenase deduced from Lactobacillus kefir (LkADH-Prince) with stereocomplementary transaminases enables the efficient deracemization of racemic aryl–alkyl secondary alcohols to the corresponding optically active α-chiral primary amines under fully aqueous, exogenous NAD(P)+-free, and highly stereocontrolled conditions, using isopropylamine as the auxiliary substrate for both biotransamination and cofactor recycling.
doi: 10.1002/cctc.70763







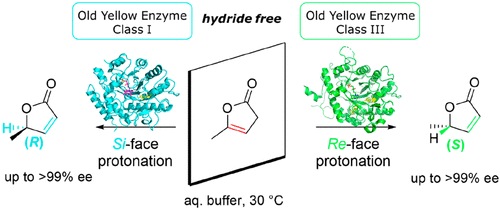
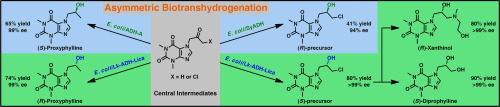
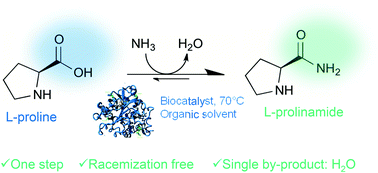
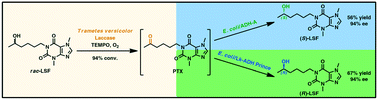
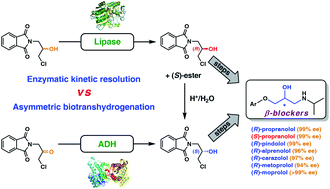
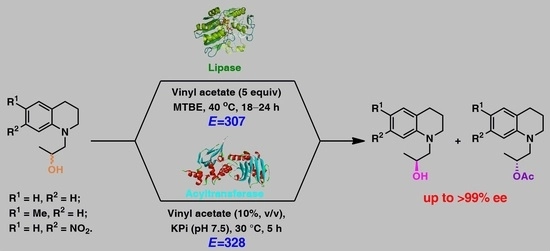
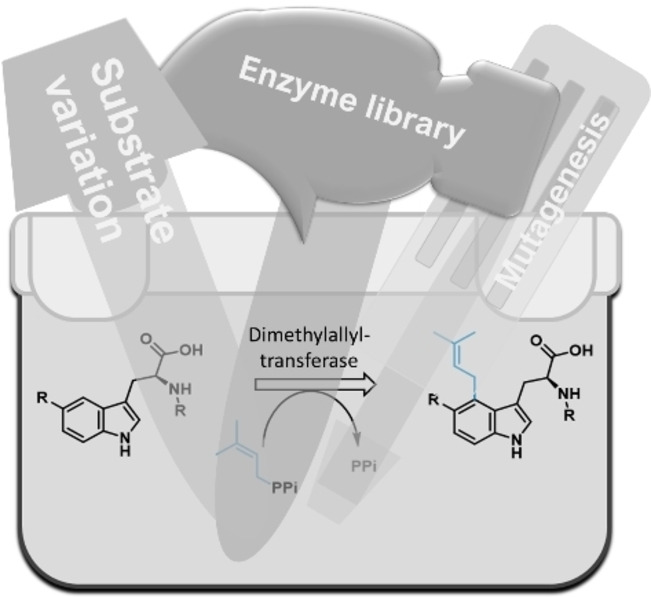
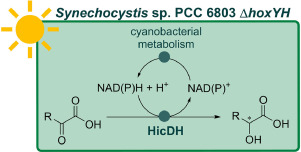

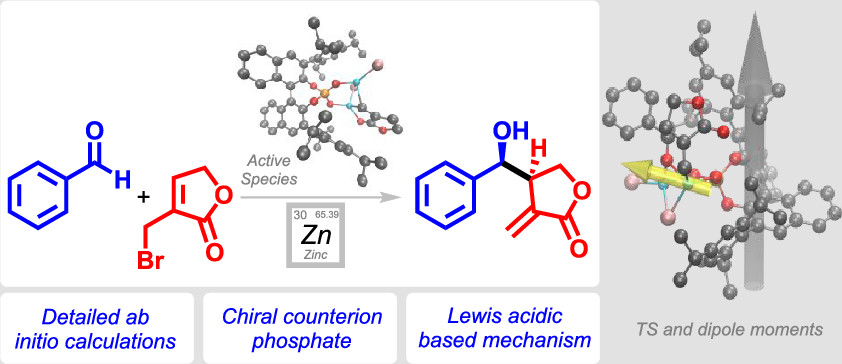
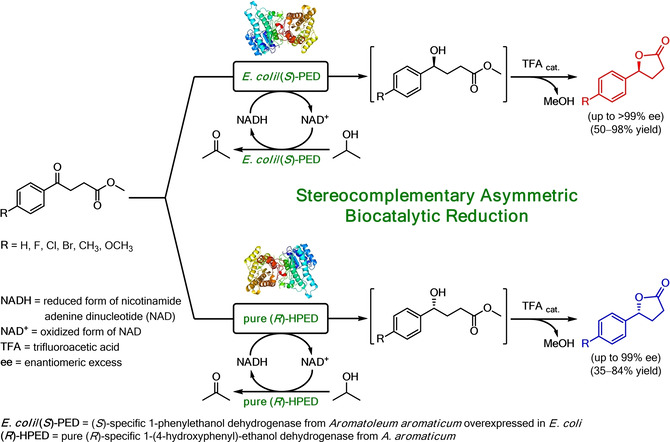
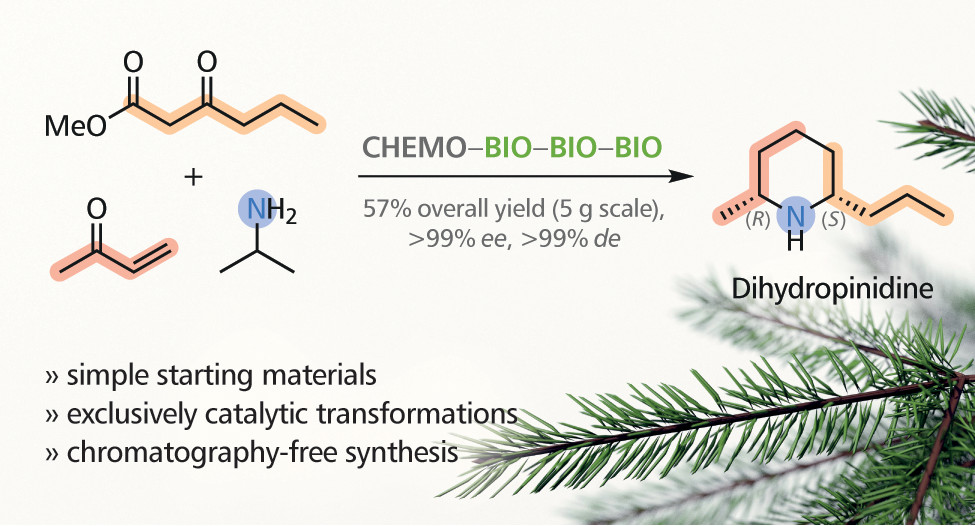
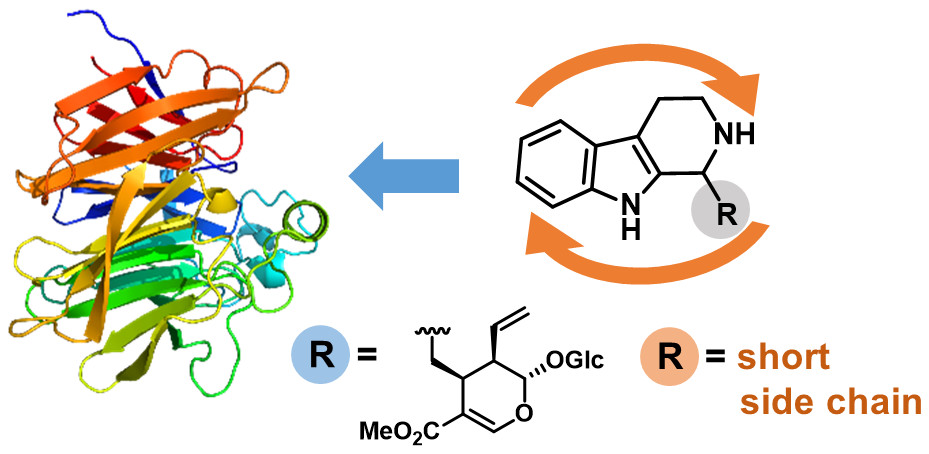




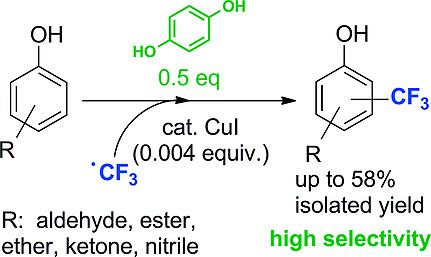
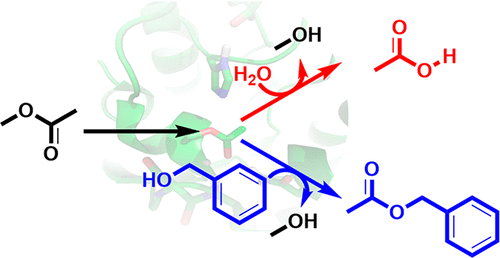
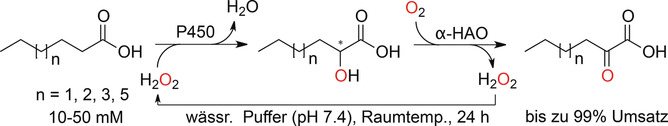
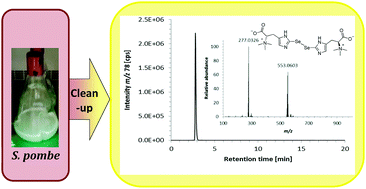
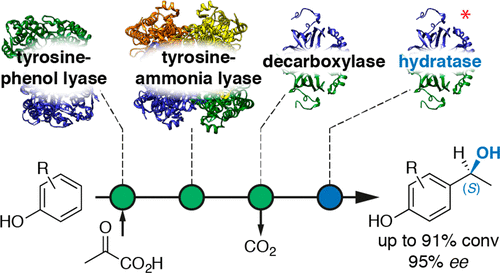
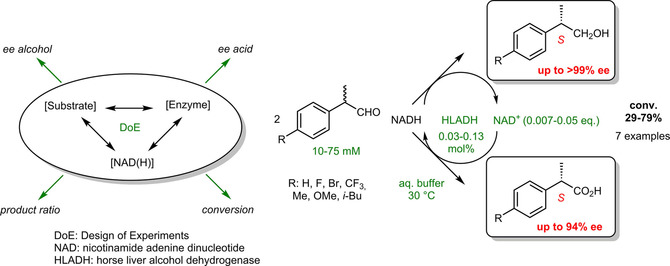
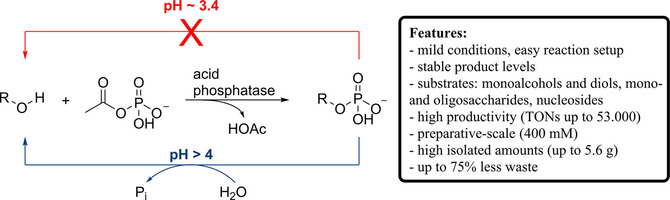
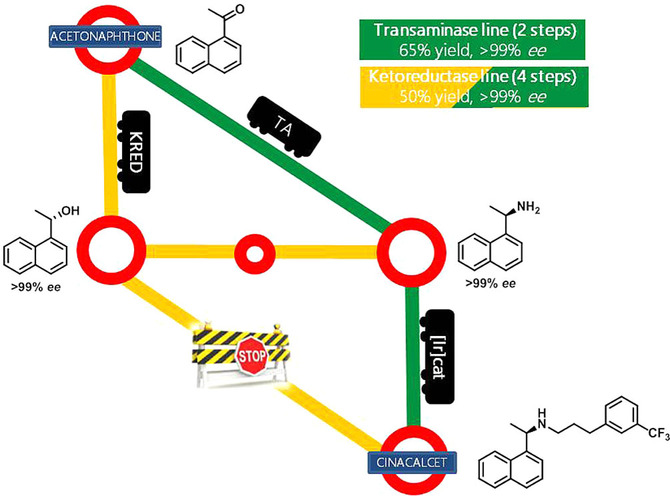

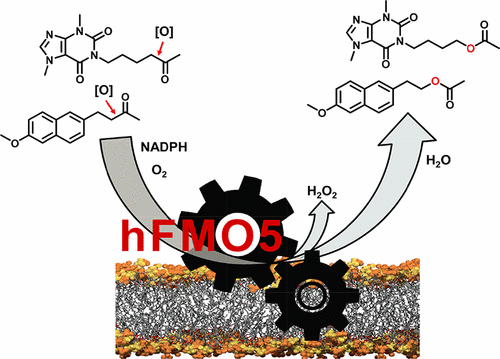
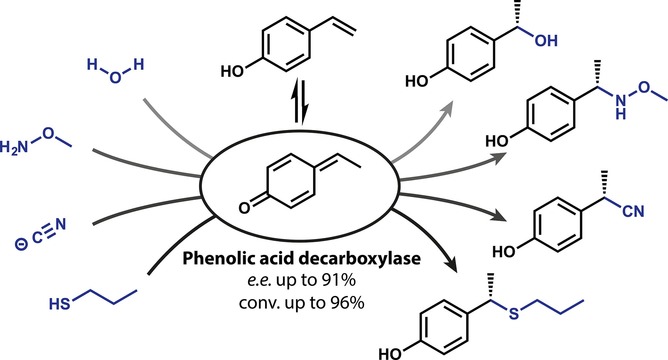
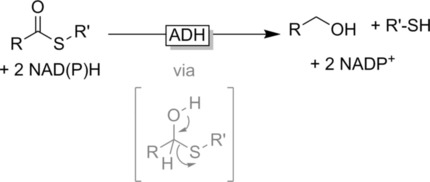
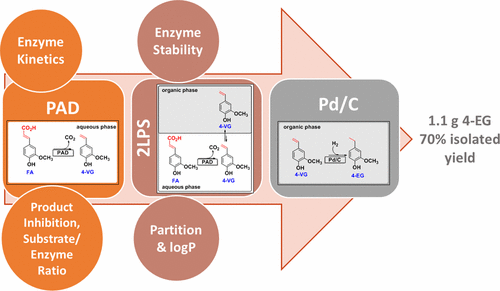

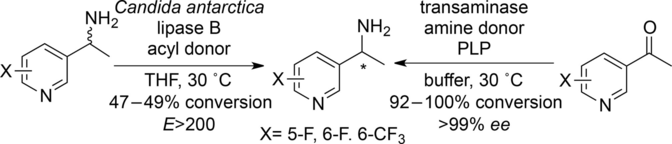
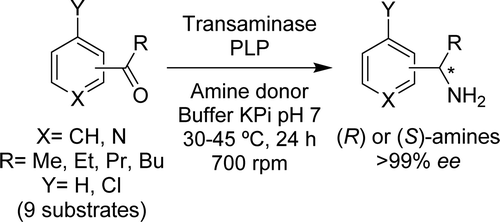
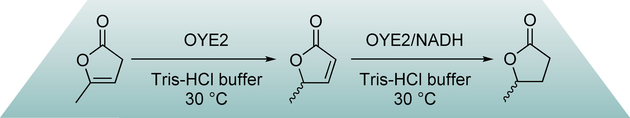
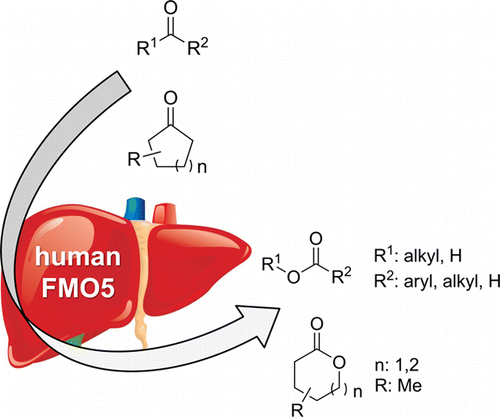
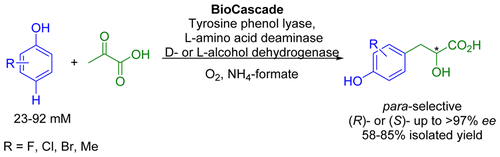
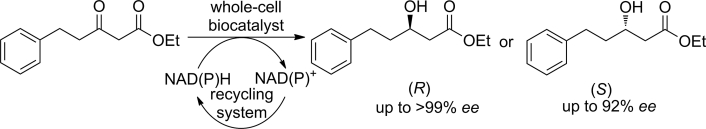
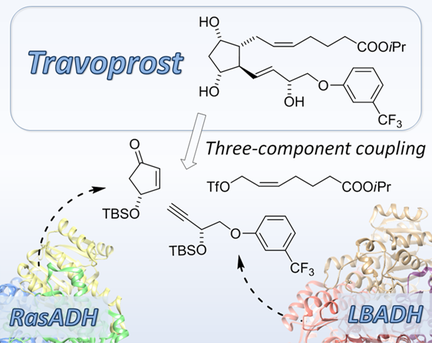
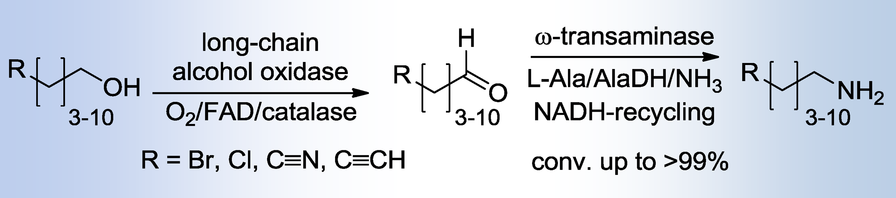
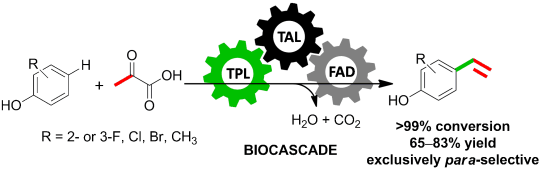
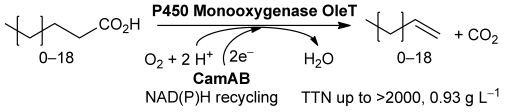




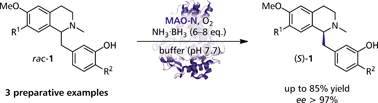







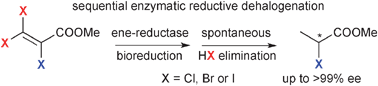
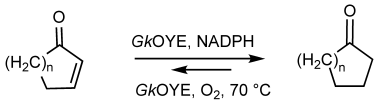
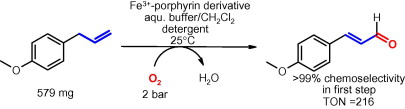
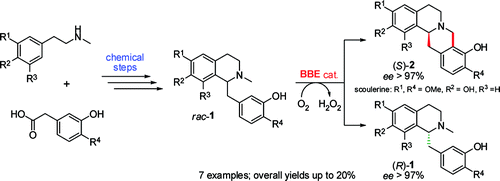
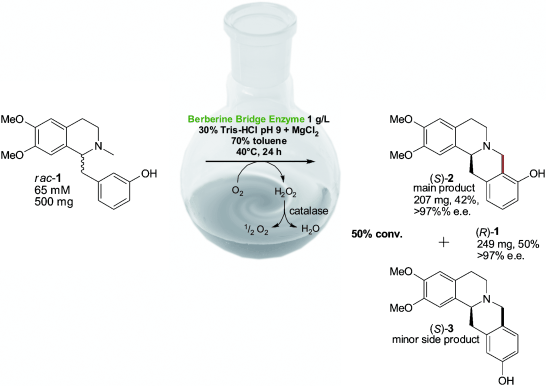


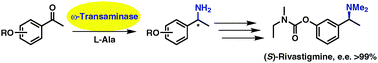

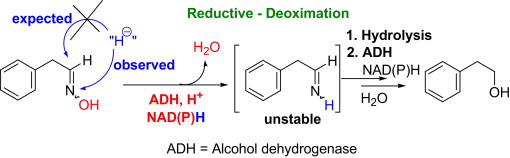
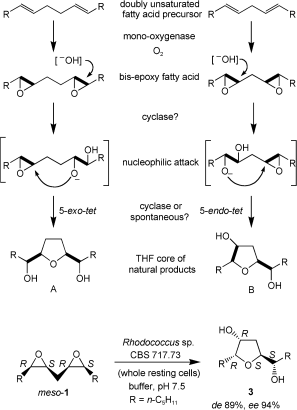
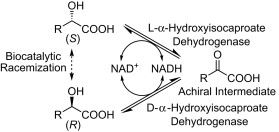
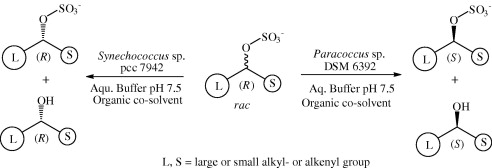
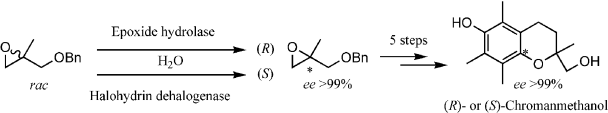









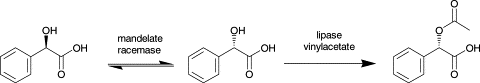
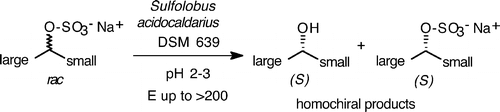
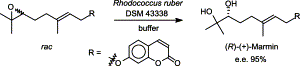
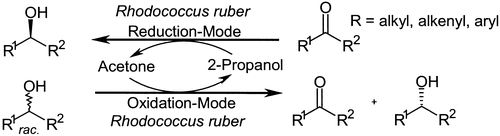

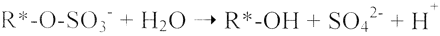
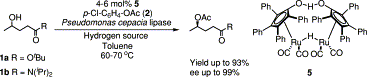
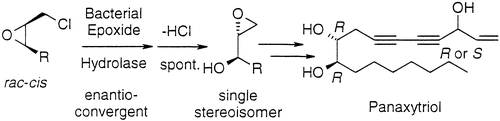
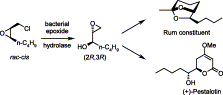
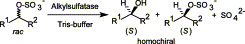
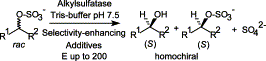

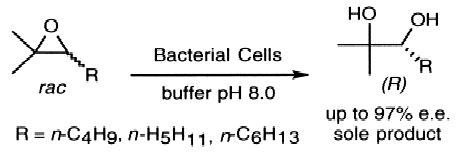
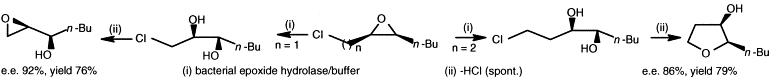
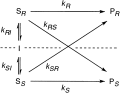

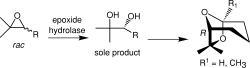
 .
.